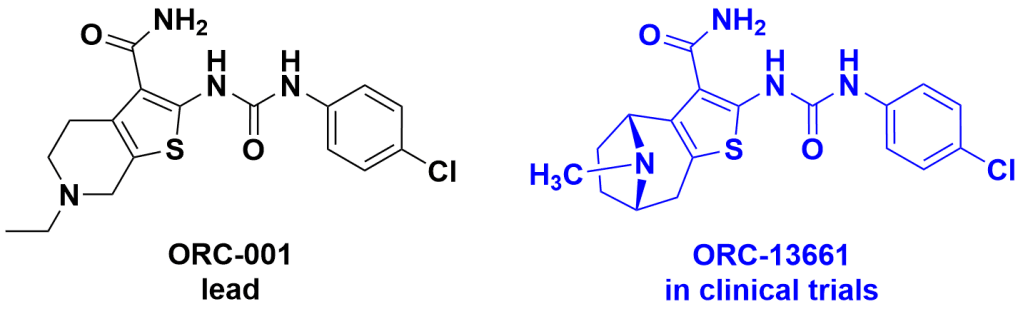
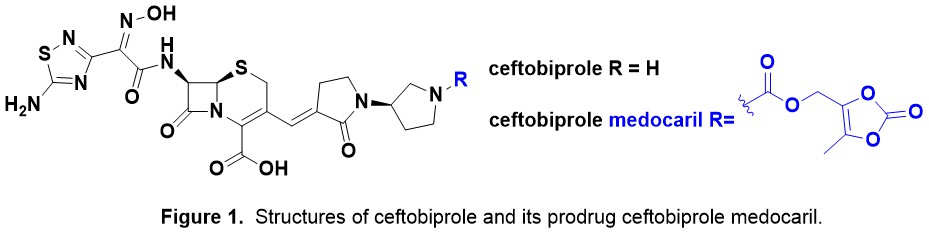
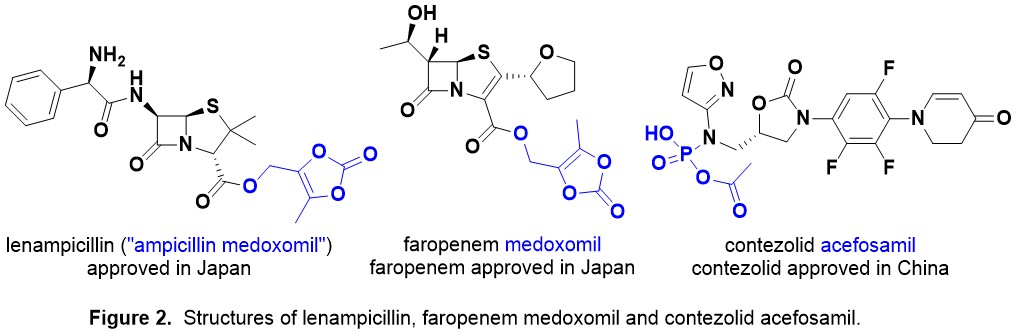
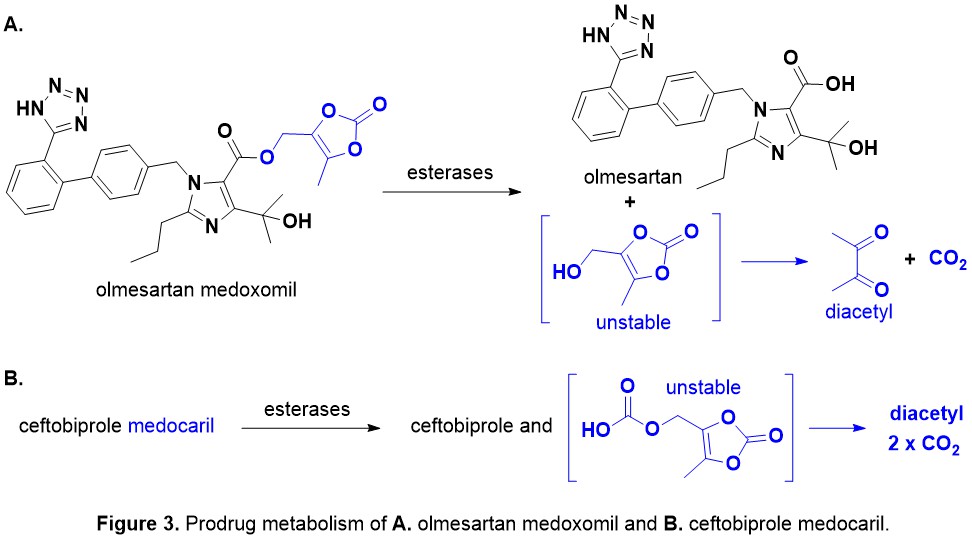
Introduction. Nature has been humanity’s pharmacy for millennia—think traditional herbal remedies, or blockbuster drugs like morphine and quinine. However, in recent decades, drug discovery focused has on alternative lead discovery methods and engineered biologics. In our new review (January 2014–June 2025) published in Natural Product Reports we ask: what is the current role of natural-product-derived (NP-D) compounds in drug approvals and clinical pipelines?
Link: https://doi.org/10.1039/D5NP00031A
The Big Picture:
– We identified 58 NP-related drugs launched globally between January 2014 and June 2025 (45 NCEs and 13 ADCs).
– Between 2014 and 2024, of the 579 drugs approved globally (388 NCEs, 191 NBEs) we found 56 (≈ 9.7 %) could be classified as NP or NP-D drugs.
– At the end of December 2024, there were 125 NP or NP-D compounds undergoing clinical trials or in the registration phase, including 33 new pharmacophores not previously seen in approved drugs. However, only one of these new pharmacophores in active clinical development has been discovered in the last 15 years.
Key Take-aways.
(1) Despite the decline in emphasis on natural products, they still contribute meaningfully (~10 %) to approved drugs.
(2) The pipeline is active—but innovation (in terms of new pharmacophores) is slow. – To unlock future NP-derived success, renewed emphasis on bioassay-guided isolation and mode-of-action elucidation is needed.
Why it Matters. For researchers, entrepreneurs and policymakers: natural products offer chemical diversity and biological relevance that synthetic libraries often struggle to match. Our review suggests that while NP-D compounds aren’t dominating the field, they remain a valuable strategic option. Re-investing in the foundational work (isolation, mechanism, natural-source exploration) could yield the next generation of breakthrough therapeutics.
Looking Ahead:
(1) Better integration of genomics, metabolomics and modern analytics with natural product discovery.
(2) Strategic partnerships between academia, industry and natural-source repositories to accelerate NP pipelines.
(3) Encourage early-phase investment in NP scaffolds and biologically-rich extracts.
(4) Further integration of machine learning, genomics, metabolomics and modern analytics with NP discovery.
Conclusion. Nature still holds surprises—and in the era of multi-drug resistant pathogens, complex diseases and the need for new therapeutic modalities, NP-derived drugs will continue to play an important role for the foreseeable future. I hope this review spurs renewed interest, investment and collaboration across the NP drug discovery ecosystem.